固体所在金属Al/Al2Cu界面局域结构研究方面取得新进展
近期,固体所张永胜研究员课题组在金属Al/Al2Cu界面局域结构研究中取得新进展,研究发现界面处局域几何结构的畸变对溶质元素 (Si、Mg、Zn) 的偏析行为有着不可忽视的影响 (图1)。相关结果以“Effects of local geometry distortion at the Al/Al2Cu interfaces on solute segregation”为题发表在Physical Chemistry Chemical Physics期刊上。
铝基合金材料由于具有高强度、低密度等优异的综合性能而广泛应用于航空航天、交通运输、汽车工业等领域。材料中的界面对提高材料的各项性能起着至关重要的作用。Al-Cu所形成的合金在190-230℃的温度范围内会在Al中析出Al2Cu,析出相可以有效地提高铝合金的强度。相关实验发现基体Al和析出相Al2Cu之间会同时形成共格和半共格界面,溶质元素Si、Mg、Zn在Al/Al2Cu界面处表现出不同的偏析行为。然而,相应的DFT-PAW理论计算的结果(溶质偏析能)和实验观察(溶质浓度)不符,这严重影响了计算预测的可靠性。
为了有效地研究Al和Al2Cu之间的界面对掺杂原子(Si、Mg、Zn)偏析的影响,张永胜研究员课题组采用了LCAO+NCPP的计算方法,结果发现界面处 (尤其是半共格界面) 局域几何结构畸变对溶质的偏析能具有很大的影响,其结果能够很好的解释实验观察:比如图2 (c)中,LCAO+NCPP计算的Si在半共格界面两侧θiˊ和Ali处的偏析能基本相同,这与图2 (b)中实验上观察到的Si在Al和Al2Cu两侧接近的浓度相吻合;而传统的DFT-PAW计算方法 (图2(d))显示,Si在Al2Cu侧的偏析能比Al侧的低,这是由于PAW方法在计算过程中固定了晶格对称性所造成。进一步的计算发现界面处的晶格失配度也对偏析能起着不可忽略的影响(图2(e)):随着半共格界面失配度的降低,Si和Mg在半共格界面处偏析能的差别也逐渐减小,接近实验观察的结果。该理论计算结果不但很好地解释了实验现象,为今后更深入而准确地研究界面的微观结构和性质提供帮助,也为将来通过界面设计调控材料性能做了良好的铺垫。
该项工作得到了国家自然科学的基金资助,所有计算都在中科院超算中心合肥分中心完成。
全文链接:https://doi.org/10.1039/D0CP00067A
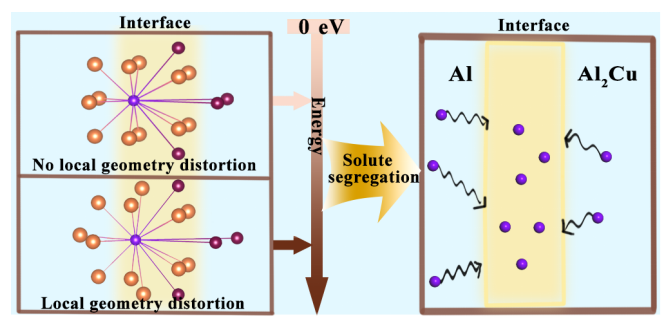
图1. 界面处几何结构畸变对溶质偏析影响的示意图。
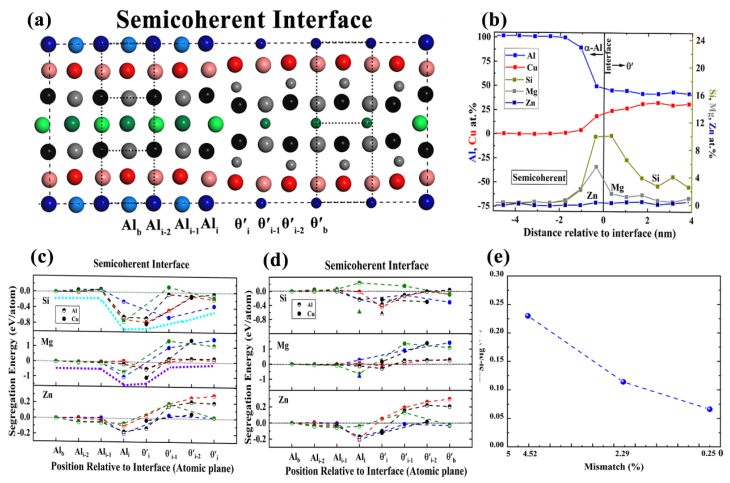
图2. (a) (010)Al||(010)Al2Cu半共格界面计算模型图中大球和小球分别代表Al原子和Cu原子,不同颜色的原子代表(c)和(d)中不同的偏析路径;(b) 实验观察的半共格界面附近Al、Cu、Si、Mg、Zn的浓度,数据来源于文章:DOI: 10.1103/PhysRevLett.105.076102;(c) LCAO+NCPP方法计算的半共格界面处溶质的偏析能,虚线代表偏析能的变化趋势;(d) DFT-PAW方法计算的半共格界面处溶质的偏析能;(e) 溶质Si和Mg偏析能之间的差值随半共格界面失配度的变化。




